Cystische fibrose is een aangeboren metabole ziekte. Lichaamsvloeistoffen zoals speeksel, bronchiaal slijm of pancreasafscheidingen zijn veel moeilijker dan normaal vanwege genetische aanleg. Gevolgen zijn onder meer ademhalingsproblemen en indigestie. Cystische fibrose is niet te genezen. Met consistente therapie kan het verloop van de ziekte echter worden vertraagd. Lees hier welke symptomen cystische fibrose veroorzaken en hoe deze te behandelen.

Cystische fibrose: kort overzicht
- beschrijving: erfelijke metabole ziekte, die taaie slijmvorming in de longen en andere organen veroorzaakt
- symptomen: Ademhalingsproblemen, irriterende hoest, longinfectie, falen om te bloeien, indigestie, ernstige diarree, leververvetting, verminderde vruchtbaarheid
- oorzaken: Overerving van defecte genen die de consistentie van lichaamsvloeistoffen beïnvloeden, uitbraak van de ziekte alleen als beide ouders een ziek gen erven (dominante recessieve overerving)
- diagnose: Bloedtest voor immunoreactieve trypsine (IRT), pancreatitis-geassocieerd eiwit (PAP), zweet test, genetische test
- behandeling: mucolytische middelen, bronchusverwijders, inhalatie, antibiotica voor infecties, cortison, CFTR-modulatoren, longtransplantatie
- voorspelling: niet te genezen, natuurlijk sterk afhankelijk van de ernst en timing van de diagnose, verkorte levensverwachting
Cystische fibrose: beschrijving
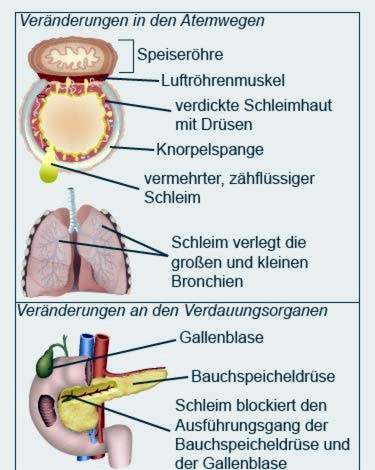
Cystische fibrose (ook wel cystische fibrose genoemd) is een erfelijke metabole ziekte. De vorming van verschillende lichaamsvloeistoffen is verstoord. De secreties van de longen, pancreas en andere organen zijn viskeuzer dan bij gezonde mensen.
Taai slijm
Het taaie slijm verstopt onder andere de kleine takken van de bronchiën en de kanalen van de interne organen. Ademhaling en spijsvertering worden vooral beïnvloed. In de loop van de ziekte kunnen de organen slechter en slechter werken.
Fout in het genetische materiaal
De oorzaak van de ziekte is defecten in het genetische materiaal. Cystische fibrose is daarom niet te genezen. Het tijdstip van diagnose en de ernst van de symptomen kunnen sterk van persoon tot persoon verschillen. Bij veel kinderen heeft cystische fibrose een enorme impact vanaf de geboorte, in andere gevallen wordt het later herkend.
Cystische fibrose: symptomen
Symptomen van cystische fibrose kunnen sterk verschillen van patiënt tot patiënt. De ziekte beïnvloedt de functie van verschillende organen, maar vooral de longen en het spijsverteringsstelsel.
Herken vroege tekenen
De eerste symptomen van taaislijmziekte variëren individueel. In de meeste gevallen verschijnen symptomen van cystische fibrose binnen het eerste levensjaar. Zo kan de ziekte meestal vroeg worden gediagnosticeerd en snel worden gestart met een therapie. Sommige patiënten hebben echter alleen significante klachten in de adolescentie. Niet elke getroffen persoon vertoont alle mogelijke symptomen. De ernst van de symptomen varieert ook.
Gewijzigde lichaamsvloeistoffen
Bij cystische fibrose is de vorming van de zogenaamde chloride-ionkanalen in de cellen verstoord. Dit verandert de samenstelling van lichaamsvloeistoffen. De eenvoudigste manier om deze verandering in het zweet van de getroffenen te detecteren. Je zweet is zouter dan gezonde mensen. De zouten natrium en chloride, die behoren tot de zogenaamde elektrolyten, zijn verrijkt met hun zweet. Als gevolg van zweten verliezen patiënten met cystische fibrose in toenemende mate lichaamszouten.
Gevarieerde symptomen van cystische fibrose
De ziekte treft een hele reeks orgaansystemen. Vaak verschijnen de eerste symptomen van cystische fibrose in de longen en het spijsverteringskanaal. In de loop van het leven kunnen verdere klachten worden toegevoegd. Door gerichte therapie kunnen de symptomen goed worden behandeld. De symptomen kunnen echter ook dreigende proporties bereiken. Het is vooral gevaarlijk wanneer de bronchiën door het viskeuze slijm verstoppen. Dan kunnen de patiënten in extreme gevallen stikken.
Cystische fibrose: symptomen van de long
Ademhalingsproblemen en irriterende hoest
In de meeste gevallen komen symptomen van de longen bij cystische fibrose alleen voor bij iets oudere baby’s. Pasgeborenen hebben meestal geen ademhalingsproblemen. Symptomen van cystische fibrose worden vaak uitgedrukt in de vorm van een hoest-keelachtige, chronische, irriterende hoest bij iets oudere kinderen. Het slijm in hun luchtwegen is verhoogd, taai en viskeus. Dit belemmert de luchtstroom in de longen. Na verloop van tijd ontwikkelt zich een progressief ademnood.
Frequente infecties
Verhoogde slijmproductie in de longen maakt het gemakkelijker voor bacteriën om te koloniseren en infecties te veroorzaken. Terugkerende longontsteking of bronchiale infecties worden voornamelijk veroorzaakt door bacteriën zoals stafylokokken en Pseudomonas-soorten. De verstoorde zoutbalans in de longen belemmert ook de afweer van het lichaam. Ook kunnen longbloedingen optreden. Een typisch teken hiervan is het ophoesten van met bloed gemengd slijm.
Hoewel de longen vanaf jonge leeftijd worden beschadigd, zijn de eerste symptomen van cystische fibrose in de luchtwegen vaak alleen op de basisschoolleeftijd of zelfs later. De symptomen treden soms alleen op wanneer grote delen van het longweefsel al zijn vernietigd of de luchtwegen ernstig vernauwd zijn.
Cystische fibrose: symptomen van de alvleesklier
Bij patiënten met cystische fibrose raakt de alvleesklier vaak ontstoken. Het scheidt een afscheiding af die onder andere enzymen bevat voor de vertering van vet en suiker. Bij mensen met cystische fibrose condenseert de secretie vanwege zijn viscositeit terug en veroorzaakt ontsteking.
Naarmate het proces vordert, wordt het pancreasweefsel verhard en met littekens bedekt. Artsen spreken van fibrose. De fibrose vernietigt geleidelijk de alvleesklier. Naast de gal vormt de alvleesklier ook insuline, die nodig is, onder andere voor het suikergebruik in het lichaam. Patiënten die wat ouder zijn (vanaf de adolescentie) ontwikkelen vaak diabetes mellitus.
Cystische fibrose: symptomen van gal
De alvleesklier en de galblaas delen een gemeenschappelijk kanaal in de darm. Daarom kan de terugstroom van alvleesklierafscheidingen ook een ontsteking van de galblaas veroorzaken. Vaak vormen zich galstenen, die de galblaas volledig van de galblaas kunnen blokkeren.
Cystic fibrosis: symptomen van het spijsverteringskanaal
Naast klachten aan de longen hebben symptomen van cystische fibrose vooral invloed op de spijsvertering. Door het gebrek aan gal is bijvoorbeeld de vetvertering aangetast. Vaak verdragen patiënten vet voedsel slecht. Het ingenomen voedsel wordt grotendeels opnieuw onverteerd uitgescheiden. Typisch zijn dan zeer volumineuze en zachte stoelgang.
Diarree en groeistoornissen
Getroffen kinderen, inclusief zuigelingen, lijden vaak aan ernstige diarree. Hoewel ze goed drinken en eten, nemen ze nauwelijks toe. Groeistoornissen en ondervoeding zijn daarom verdere klassieke gevolgen van de ziekte.
Dergelijke klachten op het gebied van spijsvertering kunnen echter ook bij andere ziekten voorkomen. Alleen in combinatie met ademhalingsproblemen zijn ze daarom een karakteristieke indicatie van cystische fibrose, die zeker moet worden onderzocht.
anale prolaps
In het verdere verloop kan cystische fibrose verschillende complicaties in het spijsverteringskanaal veroorzaken. De meest voorkomende is een zogenaamde anale verzakking. Bij anale verzakking puilt het darmslijmvlies uit de anus. Een dergelijk incident moet zo snel mogelijk chirurgisch worden behandeld.
darmobstructie
Betrokkenheid van de darm (invaginatie) of darmobstructie (ileus) komt ook vaak voor. Beide complicaties zijn geassocieerd met ernstige buikpijn en aanzienlijke spijsverteringsproblemen. De pijn treedt meestal op bij spatten, vooral na het eten. Een darmobstructie is dodelijk als deze niet wordt behandeld. Krampachtige, acute buikpijn moet daarom altijd door een arts worden opgehelderd.
Cystische fibrose: symptomen van de lever
leververvetting
Onder de terugstroom van gal lijdt ook de lever. Bij veel patiënten ontwikkelt zich leververvetting in de loop van de ziekte. Vermoeidheid, verlies van eetlust, opgeblazen gevoel en winderigheid en, in zeldzame gevallen, kunnen gevoelens van druk of pijn in de bovenbuik optreden.
levercirrose
In zeldzame gevallen ontwikkelt zich een krimpende lever (levercirrose) waarbij de lever ernstig wordt gestoord in zijn functie. Het manifesteert zich eerst in de vorm van geelzucht (geelzucht). Tekenen van geelzucht zijn de geelachtige verkleuring van wit in de ogen. Na een lange tijd treden ook hartproblemen op en de prestaties van de getroffenen blijven afnemen.
Cystische fibrose: verminderde vruchtbaarheid
Meer dan de helft van alle mannelijke patiënten is onvruchtbaar. Hoewel ze in de meeste gevallen vruchtbaar sperma kunnen vormen, kunnen ze niet door de zaadleider komen omdat ze worden geblokkeerd door viskeus slijm.
Getroffen vrouwen zijn meestal minder vruchtbaar. Over het algemeen kunnen ze een kind ontvangen en afleveren. In hun eileiders hoopt zich echter taai slijm op, waardoor het sperma nauwelijks kan doordringen. Vooral op hogere leeftijd neemt de kans op zwangerschap snel af.
Cystische fibrose: symptomen bij kinderen
Cystische fibrose is een genetische ziekte. Het is altijd aanwezig geweest sinds de geboorte. Maar de klassieke symptomen van cystische fibrose komen niet altijd voor in de kindertijd. Er zijn echter vaak al niet-specifieke instructies die moeten worden gevolgd. Dit is met name het geval als zich in de familie al gevallen van cystische fibrose hebben voorgedaan.
Opgezette buik
Een indicatie dat er een stofwisselingsstoornis is, is bijvoorbeeld een opgeblazen buik voor een lange tijd. Vaak lijden de kinderen aan diarree. Bij pasgeborenen kan een aanzienlijk vertraagde eerste ontlasting (Kindspech) een indicatie zijn van cystische fibrose. In veel gevallen treden groei- en groeistoornissen op, hoewel de kinderen met hunkeren naar eten. Alleen in zeldzame gevallen leidt het ook tot constipatie (constipatie) als gevolg van de ziekte.
Rammelende ademhaling
Andere symptomen die kunnen wijzen op cystische fibrose zijn onder meer ratelende ademhaling en ernstige rusteloosheid. Veel kinderen lijden aan chronische ontsteking van de sinussen. Deze zijn vooral merkbaar door niet bepaald lokaliseerbare pijn in het gezicht. Neuspoliepen komen vaker voor bij kinderen met cystische fibrose dan bij gezonde kinderen.
Als kinderen langer last hebben van ademhalingsproblemen of indigestie, moet altijd een arts worden geraadpleegd als voorzorgsmaatregel. Bij kinderen kunnen levensbedreigende situaties snel optreden, omdat zij zelf hun klachten niet kunnen verwoorden of hun ernst niet kan inschatten.
Cystische fibrose: oorzaken en risicofactoren
Cystische fibrose wordt veroorzaakt door een genetisch defect. De pathologische verandering ligt op het zevende chromosoom in het zogenaamde CFTR-gen.
Het CFTR-gen (cystische fibrose transmembraan regulator-gen) bevat de constructiehandleiding voor een kanaal waardoor chloride-ionen de cellen binnenkomen. De defecte chloride-ionkanalen blokkeren het transport van zout naar bepaalde lichaamscellen bij patiënten met cystische fibrose.
De aangetaste kliercellen maar in plaats van de anders vloeibare secretie komt taai slijm uit. In de longen, de neusbijholten, de alvleesklier, in de darm, in de galwegen en in de geslachtsklieren, worden viskeuze slijmafscheidingen met een hoog zoutgehalte gevormd.
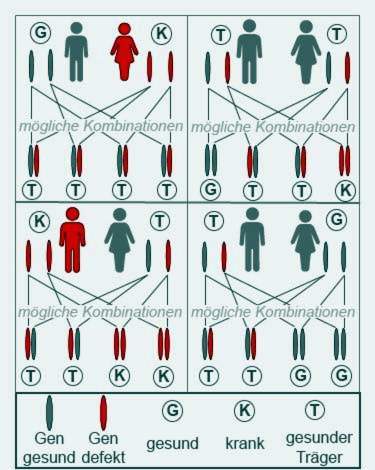
Cystische fibrose: hoe bedreigd is mijn kind?
Cystische fibrose breekt alleen uit wanneer beide ouders een pathologisch veranderd gen doorgeven aan hun kind. Deze ouders zijn dan meestal allebei gezond, maar drager van het gen.
Mensen met cystische fibrose zijn slechts gedeeltelijk vruchtbaar. Sommige patiënten worden nog steeds ouders. Zieke vaders of moeders geven echter altijd een ziek gen door, aangezien beide CFTR-genen de informatie over cystische fibrose bevatten. Hun kinderen worden echter alleen ziek als ze ook een ziek gen van de andere ouder krijgen.
Paren wiens families al gevallen van cystische fibrose hebben gehad, moeten genetisch advies inwinnen voordat ze een zwangerschap plannen.
Pre-implantatie genetische diagnose
Ouders die cystische fibrose kunnen veroorzaken, kunnen pre-implantatie genetische diagnose ondergaan. Bij genetische testen vóór implantatie worden de eicellen eerst kunstmatig bevrucht. De eerste celdelingen vinden plaats in de reageerbuis (in vitro).
Voordat een embryo wordt ingebracht, wordt het getest op veranderde geneigenschappen. Vervolgens worden alleen embryo’s geïmplanteerd die het zieke gen niet dragen. Ongeacht dit, kan ook tijdens de zwangerschap worden onderzocht of het kind later CF zal ontwikkelen.
Cystic fibrosis: onderzoeken en diagnose
In tegenstelling tot enkele jaren geleden ondergaan de meeste ziekenhuizen tegenwoordig routinematig neonatale screening. Dit omvat onderzoeken naar cystische fibrose.
Screening in bloed en zweet
Voor de screening wordt bloed afgenomen van de pasgeborene. De cystische fibrose-test bestaat uit verschillende fasen:
bloedonderzoek
Test op verhoogde niveaus van immunoreactief trypsine (IRT) en pancreatitis-geassocieerd eiwit (PAP). In geval van afwijkingen vindt de zweetproef plaats.
lasproef
Patiënten met cystische fibrose hebben een aanzienlijk hoger zoutgehalte in zweet dan gezonde mensen. Voor de cystische fibrose-transpiratietest wordt het gehalte aan natriumzouten en chloride in het lichaamszweet gemeten. Bij kinderen wordt het zweet op de onderarm verzameld en vervolgens geanalyseerd. Als hier een vermoeden ontstaat, wordt een genetische test uitgevoerd.
genetische test
Bij patiënten met cystische fibrose is het zogenaamde CFTR-gen, dat de blauwdruk voor specifieke ionkanalen biedt, veranderd. Deze constructiehandleiding is lang, het bestaat uit ongeveer 6500 basenparen. Overal kan een fout in code sluipen, maar de fouten hebben verschillende effecten. Daarom worden alleen de meest voorkomende afwijkingen in de code getest.
Familiegeschiedenis geeft hints
Als er geen geschikte neonatale screening is uitgevoerd en de vermoedelijke cystische fibrose later opkomt, is de huisarts of internist de juiste persoon om contact op te nemen. In een eerste gesprek registreert deze persoon de medische geschiedenis (anamnese). Bij vermoede cystische fibrose wordt bijzondere aandacht besteed aan de familiegeschiedenis.
Lichamelijk onderzoek
Vervolgens vindt een lichamelijk onderzoek plaats. De arts luistert naar de longen en scant de interne organen. Hij kan al enkele andere aandoeningen uitsluiten die worden geassocieerd met symptomen die vergelijkbaar zijn met die van cystische fibrose.
Bovendien kunnen röntgenonderzoek ademhalings- en longblokkades vertonen. Laboratoriumonderzoek geeft aanwijzingen voor functionele beperkingen van de interne organen. Zelfs bij volwassenen die worden verdacht van taaislijmziekte, biedt een zweettest belangrijk bewijs voor de diagnose.
Familieleden testen
Als cystische fibrose in een familie wordt gedetecteerd, is het logisch dat alle andere gezinsleden ook een onderzoek ondergaan. Cystische fibrose kan ook voorkomen in verzwakte vormen. Het duurt vaak nog vele jaren voordat cystische fibrose zich manifesteert met duidelijke symptomen. Desalniettemin is een vroege diagnose en cystische fibrose-therapie ook belangrijk voor de getroffenen om hun levensverwachting te verhogen.
Cystische fibrose: behandeling
Cystische fibrose is niet te genezen. Kinderen geboren met cystische fibrose lijden hun hele leven lang aan de gevolgen van de ziekte. Een combinatie van fysiotherapie, medicatie en inhalaties kan de progressie van de ziekte echter aanzienlijk vertragen. Een cystische fibrose-therapie moet daarom in de eerste plaats bereiken dat de getroffenen een zo normaal mogelijk leven kunnen leiden.
Leren leven met de ziekte
Vooral voor kinderen is het belangrijk om op de lange termijn met de ziekte te leren omgaan. Kinderen met cystische fibrose moeten zo vroeg mogelijk leren wat de ziekte betekent en hoe deze het lichaam beïnvloedt. Hier kan een ziekenhuisverblijf met speciale trainingseenheden nuttig zijn. Tijdens het proces leren kinderen en ouders hoe ze zichzelf moeten voeden, hoe sport eruit ziet en hoe ze zich het beste gedragen in kritieke situaties.
Hulp voor de longen
Er zijn verschillende opties om de symptomen te behandelen. Afhankelijk van de leeftijd van de patiënt en de ernst van de symptomen, worden verschillende benaderingen aanbevolen.
Mucolytische middelen
Patiënten met cystische fibrose hebben het meest last van longproblemen. Door regelmatig in te ademen met speciale additieven (mucolytica) lost het viskeuze slijm op en kan het gemakkelijk worden opgehoest.
Bronchiale dilatatiemiddelen
Zogenaamde beta-2-sympathomimetica verhogen ook de bronchiën, wat bovendien de ademhaling vergemakkelijkt.
Antibiotica tegen bacteriën
Vanwege de slechte ventilatie van de longen hebben mensen met cystische fibrose veel vaker last van bacteriële infecties van de luchtwegen. Anderzijds helpen antibiotica die op tijd worden toegediend. In sommige gevallen is het zinvol om deze permanent in te ademen.
Ontstekingsremmende medicijnen
Bij veel patiënten zijn de luchtwegen vaak of chronisch ontstoken. Dan helpen ontstekingsremmende medicijnen zoals cortison.
CFTR modulators
Ondertussen zijn de eerste medicijnen ontwikkeld die de verminderde functie van de ionkanalen verbeteren. Ze werken echter alleen voor bepaalde mutaties en dus alleen voor een klein deel van de patiënten. Hun effectiviteit is ook beperkt. De verbetering van de medicijnen wordt intensief onderzocht. Voor het eerst begonnen ze met de oorzaak van de ziekte in plaats van alleen met de symptomen.
Longtransplantatie – de laatste hoop
Voor ernstige gevallen is een longtransplantatie ook een optie. Hoe drastisch deze stap in eerste instantie ook lijkt, veel patiënten kunnen daarna een aanzienlijk lager belast leven leiden.
Voer correct in cystic fibrosis
Omdat cystische fibrose ook de spijsvertering verstoort, moeten patiënten goed op hun dieet letten. U moet de voorkeur geven aan een dieet met veel eiwitten en koolhydraten.
Er zijn ook vitaminesupplementen en mineralen. Deze laatste vervangen de zouten die de patiënten in grote hoeveelheden zweten. Omdat de alvleesklier niet goed werkt, krijgen kinderen spijsverteringsenzymmedicijnen als aanvulling op maaltijden.
Waarnemen vaccinaties
Vaccinatie is vooral belangrijk voor patiënten met CF. Bij hen spelen bacteriën gemakkelijker en worden ze vaak ernstiger dan patiënten die niet zijn voorgeladen. Vooral vaccins tegen mazelen en pneumokokken worden aanbevolen. Bovendien moet elk jaar een griepvaccin zijn.
Cystische fibrose: ziekteverloop en prognose
Cystische fibrose wordt veroorzaakt door een verandering in het genoom en is daarom niet te genezen. Voor mensen met cystische fibrose worden de levensverwachting en kwaliteit van leven meestal aanzienlijk verminderd. Zonder therapie verslechtert de gezondheidstoestand snel en de getroffenen leven meestal niet lang.
Met tijdige en consistente therapie kan het verloop van de ziekte aanzienlijk worden vertraagd. In de tussentijd leven patiënten veel langer dan een paar jaar geleden. De gemiddelde levensverwachting voor cystische fibrose is momenteel ongeveer 40 jaar. Maar velen leven ook 50 jaar en meer met de ziekte.
Complicaties en gevolgen
Zelfs met intensieve therapie kunnen complicaties steeds opnieuw optreden bij cystische fibrose. Meestal is er acute ademnood als gevolg van slechte longventilatie. Individuele delen van de long kunnen zelfs instorten (alektase).
Vaak ontwikkelt zich chronische bronchitis of longontsteking. Champignons kunnen ook de longen aantasten.
Bovendien kunnen verschuivingen in de vloeistof- en elektrolytenbalans leiden tot shock en bloedsomloop.
Andere complicaties en gevolgen zijn:
- chronische leverziekte, vooral cirrose
- Galblaasontstekingen en galstenen
- chronische ontsteking van de alvleesklier
- verstoorde hartfunctie
- acute darmobstructie (ileus)
- Intestinale invaginatie
- ondervoeding
- Diabetes mellitus
- Beperkte vruchtbaarheid van vrouwen of onvruchtbaarheid van mannelijke patiënten
Cystische fibrose is een erfelijke ziekte, daarom is preventie niet mogelijk. Mensen met een familierisico moeten genetisch advies inwinnen als ze kinderen willen. Er wordt een genetische test uitgevoerd en er wordt onderzocht of het CFTR-gen is gewijzigd. Afhankelijk van of een of beide partners het gen dragen, kan het risico voor de nakomelingen worden berekend.
In de tussentijd is in Duitsland ook pre-implantatie genetische diagnose (PID) mogelijk bij cystische fibrose. De voorwaarde hiervoor is altijd de goedkeuring van een ethische commissie. Eicellen worden buiten de baarmoeder bevrucht en alleen embryo’s zonder de problematische cystische fibrose-genen worden gebruikt.
Verdere informatie
richtlijnen:
- S2 Consensusrichtlijn “Diagnose van cystische fibrose” van de Society for Pediatric Pulmonology (2013)
- S3-richtlijn “Longziekte bij cystische fibrose” van de Society for Pediatric Pulmonology (2013)
Ondersteuning Groepen:
- Cystic Fibrosis e.V.